Details
Contact
United States
Meeting Summary
- Introduction
-
Nancy S. Pilotte, Ph.D.
National Institute on Drug Abuse
National Institutes of Health
Rockville, MDRecent research coupling the neurobiology of reward with the neurochemical sequelae of repeated cocaine administration indicates that adaptations within the dopamine system occur but do not alone underlie the enduring aspects of drug abuse. This symposium was organized to document these changes and had three objectives. The first was to summarize the neural remodeling (anatomical or neurochemical) that occurs within the mesolimbic dopamine system, after repeated exposure to and withdrawal from cocaine, and to link these neuroadaptations to their functional consequences. As the modification of one system often leads to compensatory changes in other systems, the second objective was to identify and describe the functional changes that occur in nondopaminergic neurons at various times after the cessation of cocaine administration. The third goal of this symposium was to aid in identifying enduring changes in one or more brain systems, to suggest possible neurochemical targets for developing therapeutic interventions for the medical treatment of drug abuse.
The presentations encompassed the macroscopic visions and the microscopic details of the brain after cocaine use. Data were presented detailing where cocaine itself bound in the human and the nonhuman primate brains. In addition to the expected labeling of dopamine transporter sites in areas rich in dopamine terminals, there was appreciable binding in the orbitofrontal cortex, the hippocampus, the amygdala, and the thalamus. Within these areas, the cocaine binding was not entirely displaced when various monoaminergic transport inhibitors were used as competitors.
Using imaging techniques to help define functional changes that occur subsequent to repeated drug use and withdrawal, data were presented that pointed to a profound dopamine D2 receptor deficiency in the mesostriatal dopaminergic neurons. This deficiency was accompanied by marked reductions in glucose utilization in the orbitofrontal cortex, an area sparse in dopaminergic innervation, where cocaine is thought to bind to nondopaminergic targets. Neurons from the prefrontal cortex project to striatal targets, where they may serve as cognitive pattern generators in a manner analogous to the well-known functions of the motor pattern generators of the brainstem. In this role, cortical inputs may evaluate the cognitive aspects of stimulation, including the environmental context, reinforcement, and learning, and may eventually activate or even downregulate other brain circuits that work in concert with the ventral striatum to produce behavioral activation. An acute cocaine challenge given to animals after a period of repeated cocaine treatment plus an intervening withdrawal period produces patterns of neural activation (marked by induction of immediate early gene expression) that are different from those observed after acute administration, representing yet another example of cocaine-induced neuroplasticity.
Repeated administration of cocaine has functional consequences on both dopaminergic and nondopaminergic neurons that persist after the exposures are terminated. For example, dopamine is removed from the extracellular space by uptake processes more slowly in the nucleus accumbens than in the dorsal striatum because there are fewer transporters in the accumbens than in the striatum. In behaviorally sensitized animals, uptake of dopamine by the dopamine transporter in the nucleus accumbens is even more inefficient, and a challenge injection of cocaine results in supranormal concentrations of dopamine in the synaptic space of brain areas known to be critical in mediating the reinforcing properties of drugs of abuse. This neurochemical sensitization accompanies the behavioral sensitization.
Other data suggest that the rate of uptake may be regulated by changes in the membrane potential, and depolarization decreases the rate of uptake by the dopamine transporter. Furthermore, repeated exposure to cocaine can alter the mechanisms underlying transmitter release in response to a depolarizing agent and decreases the efficiency of sodium and calcium channels in the plasma membrane. Acutely, the cocaine-induced inhibition of monoamine uptake increases synaptic transmission and blocks the inhibitory function of serotonergic autoreceptors by prolonging the residence time of the neurotransmitter in the synaptic space. At the same time, heterosynaptic modulation (by serotonin and dopamine) of transmitters such as GABA is also enhanced. Intermittent exposures to cocaine ultimately enhance the dopamine-D1 stimulation of cAMP. One of the metabolic products of cAMP is adenosine, which has transmitter activities of its own and decreases GABA release. It seems, then, that chronic cocaine use can alter the dynamic balance between different neurotransmitter systems and can lead to enduring changes in neural control. This observation is also noticed at the systems level, where withdrawal from cocaine can alter basal neural tone such that basal extracellular dopamine and serotonin concentrations fall below the limits of detection over a long period of time.
The final area of research discussed by the participants of this symposium is that of peptides and genes regulated by cocaine. One of these, corticotropin-releasing factor or CRF, is the well-known mediator of the "stress response," by which hypothalamic CRF induces the release of ACTH from the anterior pituitary gland, which in turn elicits the output of glucocorticoids from the adrenal gland (to dampen the initiating stimulation by CRF). This peptide is also found in brain regions that have not been directly linked with the peripheral stress response, such as the locus coeruleus, and nuclei within the amygdala. Acutely, the injection of cocaine increases the extracellular concentration of CRF in the central nucleus of the amygdala; cocaine withdrawal similarly activates neurons within this region. Another family of peptides, the cocaine- and amphetamine-regulated transcription factors or CART, is found in brain regions that are implicated in the rewarding properties of drugs of abuse, as well as in centers that control other behaviors associated with satiety. Finally, cocaine administration directly induces the expression of a gene, the NAC-1, within the nucleus accumbens; animals in which antisense has been used to knock out this gene do not develop behavioral sensitization to cocaine.
The variety of targets identified through these presentations are at once encouraging and daunting. They clearly point out that cocaine produces enduring neural adaptations, not only in dopaminergic systems but also in nondopaminergic systems. The recognition of the potential interactions between these systems (and those that surely exist but have not yet been identified) suggests that neuroadaptations occur at many different levels. Each level is a potential target for the development of medical interventions, with the possibilities of preventing further use and abuse and reversing adaptations induced by cocaine. An appreciation of the intricacies and interrelationships among these factors is crucial for each of us who works in this field.
- Cocaine Targets in Primate Brain: Liberation from Prosaic Views
-
Bertha Madras, Ph.D.
Harvard Medical School and
New England Regional Primate Center
Southborough, MAAttributed to Hippocrates (470-377 B.C.), this riveting quotation is a haunting description of drug abuse and addiction:
"Men ought to know that from the brain, and from the brain only, arise our pleasures, joys, laughter and jests, as well as our pains, sorrows, griefs and fears. It is the same thing that makes us mad or delirious, inspires us with dread and fear, whether by night brings sleeplessness, inopportune mistakes, aimless anxieties, absentmindedness and acts that are contrary to habit. These things that we are suffer come from the brain when it was not healthy."
Hippocrates surmised, rightfully, that the brain was the source of pleasure and pain. What he could not envision 2,500 years ago was that, at the end of the 20th century, advanced technologies would produce drugs that mimic all the sensations that the brain produces endogenously.
The progression of drug abuse to addiction and recovery can be described in phases: the acute drug phase that produces pleasure, the addiction phase, withdrawal, and abstinence. The first part of Hippocrates' quotation refers to the initial state of drug use, when sensations are positive and incentive builds to use again. The second part of the quote corresponds to the second, third, and final stages of drug use-addiction, withdrawal, and craving. This presentation focuses on the initial phase and the initial targets of cocaine in the brain.
Accumulating evidence indicates that dopamine-containing neurons are principal targets of cocaine in the brain. Dopamine is found in neurons unevenly distributed in the brain. At least four major clusters of cells produce dopamine. Of these, the mesolimbic dopamine neurons are often implicated as the mediators of reward or reinforcement. They originate in the ventral tegmental area and project to various forebrain structures, including the nucleus accumbens and cortical regions. When dopamine is released from these projection neurons, it activates at least five subtypes of presynaptic and postsynaptic dopamine receptors. Receptor activation by dopamine is rapidly terminated by a number of processes, of which transport into the presynaptic neuron by the dopamine transporter (DAT) is one of the most significant.
After intravenous administration, cocaine accumulates in dopamine-rich regions (caudate-putamen and accumbens). In these regions, a single dose of cocaine raises the extracellular concentration of dopamine, and the rise and decline of dopamine correspond temporally to the cocaine levels in the blood and brain. The increase of dopamine is attributable to blockade of dopamine transport, which results in an inundation of dopamine in the extracellular fluid. At a molecular level, the evidence is strong. The affinities of cocaine, cocaine congeners, and other inhibitors of the DAT for binding to the DAT correlate highly with their potencies for blocking dopamine transport. The relative potencies of drugs at the DAT also correlates, albeit not as impressively, to their potencies for producing behavioral stimulation, maintaining self-administration, and engendering cocaine-like subjective effects.
How and where on the molecule do drugs such as cocaine bind to and block the DAT? Can such information lead to novel drugs to treat cocaine addiction? Is it possible to design a drug that prevents access of cocaine to the transporter but allows dopamine to be transported to the interior of the cell? Drugs targeted to the transporter may have other uses, such as in the treatment of Parkinson's disease and attention deficit disorder. Transporter research is needed to address these fundamental questions and to provide important leads for effective cocaine medications. This presentation outlines two leads, generated by this laboratory, which compel revisions of some current concepts-a liberation from prosaic views.
Prosaic View #1: The DAT Is A Protein That May Form A Channel, Structured From 12 Transmembrane Domains. Where On This Protein Do Dopamine And Cocaine Bind?
Two key components of the cocaine molecule are its amine nitrogen and its aromatic ring. Most transmitters, including dopamine, serotonin, and norepinephrine, contain an amine nitrogen in their structure. Exceptions such as a newly discovered derivative of anandamide are rare. This basic structural element has driven our models of how transmitters bind to receptors and transporters and, equally importantly, has driven drug design. The paradigm for how dopamine binds to the DAT is borrowed from the beta-adrenergic receptor model. If a highly conserved aspartic acid residue on the beta-adrenergic receptor is mutated to a neutral amino acid, the capacity of the receptor to bind norepinephrine is lost. A model was constructed that proposed that the amine nitrogen of the transmitter formed an ionic bond with the carboxylic acid residue of aspartic acid. When a similar approach was applied to the DAT, by mutating a highly conserved ASP79 on the DAT, the DAT failed to transport dopamine effectively. An analogous model evolved for the DAT, which proposed that dopamine (and cocaine) bind to the DAT by the formation of an ionic bond between the amine nitrogen of dopamine and the aspartic acid residue. Such an ionic bond would also serve to form the first point of attachment between drug and transporter. Accordingly, amine drugs must mimic actions of the native neurotransmitter.
However, we (Peter Meltzer and author) recently developed a new series of compounds, based on the tropanes, that lack an amine nitrogen. These nonamines, in which the amine nitrogen is replaced with an oxygen, bound the DAT with potencies similar to those of their parent amine analogs. Furthermore, they displayed biological activity in a number of assays. These nonamines also very potently inhibited DAT transport in vitro. Nonamine represents a new class of monoamine transporter drugs. Structure-activity relationships indicate they can be either highly selective for the DAT or relatively nonselective.
What is the binding domain of these compounds? Do they see the same acceptor sites? Do they fit the same three-dimensional space as their amine-bearing counterparts such as WIN 35,428 or the GBR series, cocaine, or mazindol? The amine-bearing dopamine transport inhibitors display similar pharmacological binding profiles unique to the DAT. In this regard, the rank order of potencies of drugs that compete for the site on the transporter labeled by these radioligands is similar. Is the binding profile of [3H]O-1059, a nonamine, similar to its amine- bearing counterpart [3H]WIN 35,428? After radiolabeling [3H]O-1059, it was found to bind to a single high-affinity site on the DAT. The binding was saturable and stereoselective. To investigate whether the three-dimensional space occupied by [3H]O-1059 was the same as the monoamine [3H]WIN 35,428, competition studies with a series of potent amine-bearing drugs were conducted. The rank order of potency of drugs binding to the dopamine transporter at the nonamine site was virtually identical to the sites labeled by [3H]WIN 35,428. It can be concluded that both monoamines and nonamines bind to the same architectural elements of the transporter. Can these compelling data support the ionic theory of ligand-transporter complex formation? They suggest otherwise. The premise is not valid that an amine nitrogen is obligatory in the structure transport inhibitors; however, it may still be necessary for the association by dopamine.
The similar pharmacological specificity of the two classes of compounds can still be accounted for by hydrogen bonding between the oxa moiety (to replace the amine nitrogen) and the transporter in a region in close proximity to the aspartic acid residue. To test this hypothesis, we replaced the oxygen with a carbon atom. This area of the molecule cannot engage in any ionic or hydrogen bonding. Surprisingly, the carbon-replaced compounds bound almost as potently and selectively as their oxa or amine counterparts. This finding implies that the capacity to block transport is embedded in the three-dimensional structure of the compound and not in the functional groups. These results strongly suggest that an amine nitrogen analogous to the amine nitrogen of dopamine is not necessary for binding to or blockade of monoamine transport. In addition, the high dopamine selectivity of several of these compounds supports the concept that ionic or hydrogen bonding is irrelevant in governing selectivity. Clearly, our current model of drug-transporter interactions requires modification.
Like cocaine, a representative nonamine, O-913, increased dopamine accumulation when measured by microdialysis. It also produced subjective effects comparable to cocaine-like compounds in drug discrimination studies. How do these compounds relate to other drugs for other receptors or transporters? There are no other comparable drugs reported for transporters. Other ligands exist that have activity at receptors but bear no amine nitrogen. These include the partial agonist and active component of marijuana, delta9- tetrahydrocannabinol; anandamide; the anticonvulsant valproic acid; and the proconvulsant picrotoxin. We must also consider that receptors can be activated by pheromones and steroid hormones, which bear no nitrogen in their structure. These compounds suggest that they are the progenitors of a new generation of compounds targeted to transporters and possibly receptors. It may be feasible to design an anticocaine medication that binds to the DAT without blocking uptake, but this series of compounds do not fulfill this requirement. Even if the amine nitrogen of a drug is removed from the structure, it can still effectively block dopamine transport.
Prosaic View #2: The Dopamine Transporter Is The Principal Target Of Cocaine.
Using PET imaging, Nora Volkow and Joanne Fowler clearly demonstrated that trace doses of cocaine bind primarily in the dopamine-rich striatum. In our laboratory, ex vivo autoradiography conducted with trace or high doses of cocaine demonstrated that the greatest accumulation of cocaine occurs in the striatum. However, cocaine also distributed to other targets in the brain. The medial prefrontal cortex, hippocampus, thalamus, and amygdala all bind cocaine even though they contain low levels of dopamine. PET imaging also reveals significant accumulation of cocaine in the orbitofrontal cortex. Are these cocaine binding sites associated with the DAT? Are they relevant to the behavioral effects and abuse liability of cocaine?
To clarify the subsequent findings, we must revisit early behavioral and binding experiments that implicated the dopamine transporter as a mediator of the behavioral effects of cocaine. The potencies of drugs for binding to the dopamine transporter are correlated with their ability to elicit self-administration. However, there is one caveat to these findings. If the potencies of DAT inhibitors that are cocaine congeners for producing cocaine-like behavioral effects are compared with their potencies at cocaine binding sites, the data yield a steep binding slope. However, noncongeners produce shallow binding slopes, implying that the noncocaine congeners are considerably weaker in vivo than in vitro at the DAT. Although pharmacokinetic considerations may account for these observations, other explanations may also be relevant.
We specifically examined areas poor in dopamine transporters and assessed the binding of cocaine congeners and noncongeners to these regions. Cocaine and its congeners bound to sites labeled by [3H]cocaine in dopamine-poor areas with an appropriate rank order of potency. However, noncocaine congeners, dopamine, norepinephrine, and serotonin did not bind to these sites in DAT-poor areas. These low-density sites were not associated with dopamine transporters. Such sites were found in the medial prefrontal cortex, the hippocampus, the amygdala, and the DAT-depleted striata of patients with Parkinson's disease. Similarly, with PET imaging, we found high accumulation and low dissociation of cocaine in the orbitofrontal cortex. Although these regions have low affinity for dopamine itself and low affinity for other transport inhibitors, they may contribute to some of the psychological and behavioral effects of cocaine, including craving and withdrawal. These targets of cocaine may contribute to the pharmacological effects of cocaine, but further studies are needed to characterize these sites. These "liberating results" compel us to reexamine some of the premises that have driven cocaine research and drug design.
Acknowledgments
The author thanks collaborators Peter Meltzer, Anna-Liisa Brownell, Susan George, Roger Spealman, Susan Amara, Mark Sonders, Randy Blakely, and Michael Fahey and acknowledges the technical assistance of Helen Panas and Keiko Akasofu and graphics production by Sandy Talbot. The research described herein was supported by National Institute on Drug Abuse Research Grant Nos. DA-06303, DA-09462, DA-11558, DA-00304, and RR-00168.
Selected References
- Canfield, D.R.; Spealman, R.D.; Kaufman, M.J.; and Madras, B.K. Autoradiographic localization of cocaine binding sites by [3H]CFT ([3H]WIN 35,428) in the monkey brain. Synapse 6:189- 195, 1990.
- Madras, B.K., and Kaufman, M.J. Cocaine accumulates in dopamine-rich regions of primate brain after i.v. administration: Comparison with mazindol distribution. Synapse 18:261 -275, 1994.
- Madras, B.K.; Pristupa, Z.B.; Niznik, H.B.; Liang, A.Y.; Blundell, P.; Gonzalez, M.D.; and Meltze r, P.C. Nitrogen-based drugs are not essential for blockade of monoamine transporters. Synapse 24:340 -348, 1996.
- Meltzer, P.C.; Liang, A.Y.; Blundell, P.; Gonzalez, M.D.; Chen, Z.; Georg e, C.; and Madras, B.K. 2-Carbomethoxy-3-aryl-8-oxabicyclo [3.2. 1]octanes: Potent non-nitrogen inhibitors of monoamine transporters. J Med Chem 40:266 1-267 3, 1997.
- The Brain is not the Same After Chronic Cocaine: Network-Level Changes in Basal Ganglia Circuits
-
Ann Graybiel, Ph.D.
Massachusetts Institute of Technology
Cambridge, MAThe ventral striatum, the ventral pallidum, and the dopaminergic and serotonergic inputs to these regions are thought to be critical to the expression of the behavioral and rewarding effects of cocaine. Such expression occurs under the modulating influence of complex mechanisms that involve cue stimuli and stimuli related to reward. One can think of such a modulating neurocircuit as consisting of the dorsally lying basal ganglia, the thalamus, and the frontal cortex. This circuit seems to act as a cognitive pattern generator (analogous to the motor pattern generators of the brainstem) that can evaluate the cognitive aspects of stimulation and eventually activate other brain circuits wired in with the ventral striatum and pallidum to produce behavioral activation. We know that the neurocircuitry of the dorsal striatum, with its dopamine inputs, falls into two broad categories connected with the striosome and the matrix compartments. We also know that this neurocircuitry is strongly linked to both the limbic system via the striosomes and the sensory-motor circuitry of the striatum via the matrix. An emphasis on the circuitry is instructive because chronic treatment with cocaine or amphetamine, in contrast to acute administration of these drugs, presents a compelling model of neuroplasticity: We believe different neural circuits become activated in response to cocaine as a result of chronic exposure to the drug. This presentation emphasizes data supporting this view for circuits involving the dorsal striatum.
The acute administration of cocaine or amphetamine induces striking increases in the expression of immediate early genes (IEGs) that serve as markers of neural activation. We assessed the activation of c-Fos, JunB, and ARC (a cytoplasmic gene related to the cytoskeleton). Acutely, cocaine activated many regions, particularly the matrix component of both the ventral and dorsal striatum. The degree of activation was not dose dependent, and we obtained similar results for a variety of genes. When we conducted precisely the same experiments using amphetamine instead of cocaine, we found that the pattern of activation was very different. We observed that, in the entire front end of the caudate-putamen, the predominant activation was in the striosomes, instead of in both compartments as had been seen after cocaine.
When cocaine is administered repeatedly, its effects are remarkably long-lasting. For example, in the rat, behavioral sensitization persists for as long as 87 days after a short period of repeated cocaine administration; it is thought that the central effects of repeated psychostimulant administration can essentially last a lifetime in an animal. Therefore, we asked what the effects of chronic cocaine treatment would be on gene induction in the striatum. We varied the number of days that rats were given cocaine and then probed for the expression of the IEGs. As others have found, after acute treatment with cocaine followed by a survival time of 2 or 18 hours, we could see the induction both of IEGs such as Fos and JunB and of chronic FRAs (Fos-related antigens). With 7 days of treatment (with 2-hour survival time withdrawal), we observed a downregulation of c-Fos and JunB and an upregulation of chronic FRAs, monitored by Western blot analysis.
When we analyzed the anatomic compartments in the caudate-putamen showing the gene activation, we were surprised to find that, after only 4 days of chronic cocaine treatment, a shift had begun to occur in the anatomic localization of the activation relative to that seen after acute cocaine or amphetamine treatment. We found that the IEG expression shifted toward a striosomal pattern, which is what we had seen with acute amphetamine treatment but not with acute cocaine treatment. We traced changes in levels of expression in these gene products over the course of 7 days, when Fos and JunB were clearly downregulated. But what happened during withdrawal of cocaine? We plotted the recovery of gene expression to a challenge by giving an injection of cocaine after different lengths of withdrawal and measuring how much IEG induction occurred. After a week of withdrawal that followed a week of repeated cocaine treatment, the previously suppressed c-FOS had recovered by at least half. There was a striking lateral and anterior pattern of patchiness of JunB and of FRAs in the striatum. Even more important, when the Fos response reappeared, it appeared heightened in the striosomes, with much less expressed in the matrix. These results suggested that we were seeing a change in the inducibility of genes and the expression of their gene products that had some real circuit pattern to it-possibly a very important aspect of the effects of chronic exposure to cocaine. Our results suggest that the brain circuits responding to the drug after chronic use are different from the circuits responding to the first acute dose.
Double-staining Fos-expressing cells for dynorphin showed that most responding (Fos-positive) cells were dynorphin-positive. These findings show an interesting correlation with those of Yasmin Hurd and Miles Herkenham, who published a single-case report of striatal prodynorphin mRNA in situ hybridization in a sudden-death cocaine user. They found a marked increase of prodynorphin in striosomes, relative to control levels. Thus, both in this (single) human case and in our animal studies, the results suggest that cocaine tends to activate the striosomal system after chronic use.
In primates, the only regions known to project selectively to striosomes are the anterior part of the anterior cingulate gyrus (or caudal medial prefrontal cortex) and the caudal orbital frontal cortex. These areas are strongly implicated in psychiatric disorders such as obsessive-compulsive disorder. These regions of the cortex are tightly linked to the amygdala, the hippocampus, and the mediodorsal thalamus by circuits involved in learning and memory and to limbic circuits that relate, for example, to the locomotor behavior mediated by the nucleus accumbens.
This neural circuit, linked to the striosomal subsystem within the striatum, in certain ways resembles the ventral tegmental area in that nondopaminergic cells appear to enjoy a special relationship with dopamine-containing neurons of the midbrain. These special striatal compartments may instruct the reward-related neurons of the nigral/ventral tegmental area complex. One of the things that may occur at the systems level by virtue of repeated exposure (of an animal or person) to cocaine is some shift in the normal balance between the context and evaluation of stimuli, heavily influenced by reward signals.
Dopamine and glutamate are key coplayers in many neuroplastic systems within the basal ganglia. Glutamatergic cortical afferents project to the striatum, perhaps bearing presynaptic dopamine receptors. They synapse on the spines of the medium spiny neurons. Many dopamine-containing afferents terminate on the very same spines that receive cortical inputs, suggesting that dopamine can fine-tune the inputs to the spiny neurons. Both dopamine and glutamate strongly affect plasticity in the corticostriatal system, perhaps by LTP and LTD. Therefore, we wanted to be able to look at the effects of dopamine on glutamate transmission in the striatum.
We stimulated the somatomotor cortex in monkeys and rats, looked for striatal activation of IEGs as a measurable population response, and found that activation of c-Fos and JunB occurred in the matrix, in projection neurons that express enkephalin. To look at the effects of dopamine on this corticostriatal system, we concentrated on studying cortical activation in the rat. We first developed a method for stimulating the cortex in awake rats. We implanted chronic wells over the somatomotor cortex and removed local GABAergic inhibition in the cortex by applying picrotoxin (or CSF, for the control studies) epidurally over the somatomotor cortex via the chronically implanted wells. We were able to activate striatal IEGs using this methodology.
To study the effects of dopamine transmission on this activation, we first systemically injected the broad-spectrum dopamine D2 antagonist haloperidol and then applied the local picrotoxin activation to the cortex. This combination produced a strong synergistic IEG activation, as if there were enhanced input signaling from the cortex when D2 receptors were blocked. Intrastriatal injections of the dopamine D2 receptor antagonist sulpiride also enhanced corticostriatal transmission as measured by gene induction. Thus, dopamine receptors inside the striatum (which are targets of cocaine) can have a dramatic effect on corticostriatal transmission. We further found that this activation of striatal IEGs was dependent on NMDA receptor activation, as it was inhibited by application of the antagonist MK801.
How would chronic cocaine exposure affect corticostriatal transmission? To study this issue, we administered cocaine or saline i.p. to rats for 1 week to downregulate Fos and then locally stimulated the cortex by epidural application of picrotoxin to see whether the cocaine exposure would influence cortically evoked gene expression in the striatum. In control experiments we found, as expected, that Fos remained downregulated after the combination of repeated cocaine plus systemic saline or after repeated cocaine plus acute cocaine. Repeated saline plus picrotoxin also produced the expected robust corticostriatal induction of Fos, but repeated cocaine led to a dramatic decrease in the ability of the corticostriatal stimulation to activate IEGs in the striatum in the repeated cocaine plus picrotoxin group. This experiment shows that it is possible to look at circuit-level changes that go beyond inducing genes merely by giving drug. We can use the cortical stimulation to probe the effects of cocaine on corticostriatal circuit function.
It is clear that cocaine is a major activator of basal ganglia loops and that cocaine can lead to massive neural activation at the circuit level. If we look at the full circuit diagram of outflow of the basal ganglia, including pathways leading to the neocortex, it is striking that the basal ganglia outflow leads not only to the somatomotor cortex or premotor cortex but also to the prefrontal cortex. This pattern of connectivity suggests that some of the circuit-level changes found after even a week of cocaine treatment could influence not only downstream circuits that involve locomotion and the development of stereotypy but also upstream circuits leading to what we have called cognitive pattern generators in the cortex. This may be one way in which cocaine can influence cognitive activity after chronic exposure to this drug.
Acknowledgment
This research was supported by National Institute on Drug Abuse Grant No. DA-08037.
Selected References
- Berretta, S.; Parthasarathy, H.B.; and Graybiel, A.M. Local release of GABAergic inhibition in the motor cortex induces immediate-early gene expression in indirect pathway neurons of the striatum. J Neurosci 17:4752-4763, 1997.
- Graybiel, A.M. Building action repertoires: Memory and learning functions of the basal ganglia. Curr Opin Neurobiol5:733-741, 1995.
- Graybiel, A.M. The basal ganglia and cognitive pattern generators. Schizophr Bull 23:459-469, 1997.
- Hillegaart, V.; Berretta, S.; and Graybiel, A.M. Effects of chronic cocaine exposure on corticostriatal transmission in the rat. Soc Neurosci Abstr 22:410, 1996.
- Hurd, Y.L., and Herkenham, M. Molecular alterations in the neostriatum of human cocaine addicts. Synapse 13:357-369, 1993.
- Moratalla, R.; Elibol, B.; Vallejo, M.; and Graybiel, A.M. Network-level changes in expression of inducible Fos-Jun proteins in the striatum during chronic cocaine treatment and withdrawal. Neuron 17:147-156, 1996.
- Changes in Human Brain Systems After Long-Term Cocaine Use
-
Nora Volkow, M.D.
Brookhaven National Laboratory
Upton, NYTo understand the biochemical changes that occur in the brains of individuals who are addicted to cocaine, we have taken advantage of nuclear medicine techniques and targeted the dopaminergic system of detoxified cocaine users for PET studies. Involvement of dopaminergic systems in reinforcement is clear, but its role in the addictive processes is much less clear. We must understand the process that occurs between taking the drug because it is pleasurable and the addictive state, during which the drug is taken whether or not it is pleasurable. In other words, we must differentiate the components of the drug that give pleasure from the components involved in compulsive drug-taking.
What is the role of dopamine in addictive behaviors? The dopaminergic system is complex and functions classically (synaptically) and tonically (baseline state), and we must approach it from both perspectives. Our investigations of this system have been made in its basal state and during a pharmacological challenge. We have tried to determine whether there are abnormalities in the dopamine synapse in cocaine addicts. We assessed the integrity of the presynaptic terminal by measuring binding of dopamine uptake inhibitors such as methylphenidate and [11C]-cocaine to the dopamine transporter. Postsynaptically, we labeled the dopamine D2 receptor. (The dopamine D2 receptor resides both presynaptically and postsynaptically; however, because PET technology has relatively poor spatial resolution and the density of postsynaptic sites is greater than that of the presynaptic sites, most of the signal derives from binding of [11C]-raclopride to the postsynaptic sites.) We also measured the rates of glucose utilization in these same subjects to assess metabolic changes that might occur with prolonged cocaine use.
What have we found? Detoxified cocaine abusers bind less [11C]-raclopride to dopamine D2 receptors in the basal ganglia than normal controls because they have fewer dopamine D2 receptors. This deficit appears to be long-lasting; it persists even as long as 4 months after detoxification. We were also concerned that repeated cocaine use might lead to neurotoxicity such as that seen after methamphetamine, thinking that if the density of transporters were reduced, these subjects might be at a higher risk for Parkinson's disease. We found that detoxified cocaine abusers have dramatic decreases in [11C]-cocaine binding compared with controls. However, when only the high-affinity component of binding at the dopamine transporter in the striatum is examined, there are no differences compared with controls. These measures are highly variable in controls. We saw no degeneration of terminals after cocaine treatment, nor was there an increase in striatal dopamine transporters, regardless of length of time since the last cocaine treatment.
What is the functional significance of having a decrement in D2 receptors? We looked to brain glucose metabolism. Detoxified cocaine addicts show markedly less metabolic activity in the frontal cortex and a limited decrease in activity in the basal ganglia. However, these are persistent deficits. We then correlated the regional cerebral glucose metabolism with the availability of dopamine D2 receptors. The strongest correlations were specific and corresponded with the dopamine projections to the orbitofrontal cortex and cingulate gyrus (projections that go to the striatum). Low densities of dopamine D2 receptors were associated with decreased metabolism in the orbitofrontal cortex, whereas metabolism in other regions with dopamine D2 receptors was relatively normal.
Previously, we showed that a massive activation of metabolic activity of these same brain regions was associated with very intense craving for cocaine. Decreased metabolic activity in the orbitofrontal cortex is also associated with obsessive-compulsive disorder (OCD). This correspondence raises many interesting questions: Is the orbitofrontal cortex involved in craving? Does cocaine-taking by addicts represent a compulsive behavior? If the orbitofrontal cortex is destroyed, can behavior no longer be self-controlled? Or will individuals with a dysfunctional orbitofrontal cortex emit repetitive behaviors that cannot be terminated? How does the addict behave under conditions that will elicit drug-taking behavior or craving? Is the orbitofrontal cortex involved in cue-elicited behavior? Does the decreased metabolic activity reflect an inability to release dopamine? And how can changes in dopamine release be measured?
We measured relative changes in dopamine accumulation secondary to the occupation of the dopamine transporters by marking postsynaptic dopamine D2 receptors with [11C]-raclopride and then giving our subjects a dopamine uptake inhibitor (not radiolabeled). We used methylphenidate to inhibit uptake, because we could not give cocaine to normal controls. Inhibiting the uptake of dopamine should increase the amount of dopamine in the synapse, which should compete with the labeled raclopride. Thus, the binding of [11C]-raclopride would be related to the number of free dopamine D2 receptors and [11C]-raclopride occupation of the dopamine D2 receptors premethylphenidate and postmethylphenidate can be compared as a measure of synaptic transmission.
The responses in both the controls and detoxified cocaine users were quite variable. Some of the variability was due to age, not pharmacokinetic differences: The most robust responses were observed in young subjects (dopamine transmission decreases with increasing age). As expected, methylphenidate produced a striking change in [11C]-raclopride binding. Interestingly, the control subjects reported a more intense "high" than did the detoxified cocaine users after methylphenidate. Normal controls self-reported more restlessness. Cocaine abusers (3-6 weeks after last cocaine) reported that the methylphenidate made them crave cocaine, whereas the controls did not report such craving.
We did not expect to see the biochemical changes we obtained. Normal controls showed marked reductions in striatal [11C]-raclopride binding after methylphenidate, but the cocaine abusers did not. In fact, their dopamine D2 binding was much lower than controls. The self-reports of "high" or craving obtained from the detoxified users were very blunted compared with normal controls, suggesting that one of the long-term effects of repeated cocaine use may be a state of relative dopamine dysfunction. Considering that dopamine may impart salience and motivation to an action, the cocaine abuser may be much less responsive to normal stimulation. A downregulation in dopaminergic activity may help explain the anhedonia reported by cocaine users, and they may be taking cocaine to reverse the dysphoria (self-medication). To understand craving and the role of dopamine, it is necessary to understand that the craving likely occurs as the result of the ability of dopamine to facilitate the activation of specific brain regions, like the orbitofrontal cortex, the hippocampus, and the striatum. In addition, it is the pattern of activation that leads to craving, not just one brain region and not just the increase in dopamine. Profound disruptions are found in the dopamine system of cocaine addicts, but the dopamine system is not, by itself, responsible for craving or for addiction.
Acknowledgment
This research was supported by National Institute on Drug Abuse Grant No. DA-06891.
Selected References
- Volkow, N.D.; Wang, G.J.; Fischman, M.W.; Foltin, R.W.; Fowler, J.S.; Abumrad, N.N.; Vitkun, S.; Logan, J.; Gatley, S.J.; Pappas, N.; Hitzemann, R.; and Shea, C.E. Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature 386:827-830, 1997.
- Volkow, N.D.; Wang, G.J.; Fowler, J.S.; Gatley, S.J.; Ding, Y.-S.; Logan, J.; Dewey, S.L.; Hitzemann, R.; and Lieberman, J. Relationship between psychostimulant-induced "high" and dopamine transporter occupancy. Proc Natl Acad Sci U S A 93:10388-10392, 1996.
- Volkow, N.D.; Wang, G.J.; Fowler, J.S.; Logan, J.; Gatley, S.J.; Hitzemann, R.; Chen, A.D.; Dewey, S.L.; and Pappas, N. Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature 386:830-833, 1997.
- Cocaine Causes Long-Term Synaptic Modulation in The Ventral Tegmental Area
-
John T. Williams, Ph.D.
Oregon Health Sciences University
Portland, ORAcute Effects Of Cocaine
The acute effect of cocaine is to act at monoamine transporter proteins to block the reuptake of 5-HT, noradrenaline, and dopamine. Through the inhibition of monoamine reuptake, cocaine has three general effects on synaptic transmission (figure 1). First, the duration of monoamine- mediated synaptic potentials is dramatically prolonged. Second, monoamine autoreceptor activation is augmented to inhibit further release of monoamines. Third, heterosynaptic modulation of nonmonoamine transmitter release mediated by presynaptic monoamine receptors is augmented. Each of these actions of cocaine has been observed in brain slice experiments using the ventral tegmental area (VTA). It is interesting to note that all of the observations made in the VTA using low concentrations of cocaine, when applied acutely, center on the inhibition of the 5-HT transporter.
Neurons originating in the VTA are thought to play a key role in the formation of addictive behaviors. Thus, the effects of cocaine on the membrane properties and synaptic potentials on neurons in this area are critical to understanding the cellular effects of this drug. There are at least three different populations of VTA neurons identified by the different synaptic potentials and the response to dopamine, 5-HT, and opioids. The majority of cells (about 60 percent) were tyrosine hydroxylase positive and were hyperpolarized by dopamine. These cells had a slow GABAb-mediated inhibitory postsynaptic potential (IPSP) and are termed "principal cells." About 10 percent of cells, termed "secondary cells," were hyperpolarized by [Met]-5-enkephalin and failed to respond to dopamine. A third group of cells, "tertiary cells," were hyperpolarized by dopamine, 5-HT, and [Met]-5-enkephalin. These cells had a slow 5-HT1a-mediated IPSP (Cameron et al. 1997).
The 5-HT1a-mediated IPSP observed in tertiary cells in the VTA was very sensitive to cocaine. Low concentrations increased both the amplitude and prolonged the duration of this IPSP. Higher concentrations of cocaine further prolonged the duration but decreased the amplitude of the IPSP. These results indicate that the inhibition of 5-HT reuptake by cocaine has a dramatic effect on 5-HT-mediated synaptic transmission. The prolonged presence of 5-HT increased the duration of the IPSP and caused a feedback inhibition of further 5-HT release by an action on 5-HT1d receptors located on the 5-HT- releasing terminals. A third acute effect of cocaine was observed while recording from principal neurons in the VTA (Cameron and Williams 1994). Cocaine caused a concentration-dependent presynaptic inhibition of the GABAb-mediated IPSP. This inhibition was mimicked by exogenously applied 5-HT and the 5-HT releaser, fenfluramine (10 M). In addition, the inhibitions by cocaine, 5-HT, and fenfluramine were all antagonized by the nonselective 5-HT antagonist metergoline (1 M). To confirm that the action of cocaine was mediated through the inhibition of reuptake of endogenous 5-HT, slices were pretreated with parachloroamphetamine (PCA, 10 M, 2-4 hr) to deplete endogenous 5-HT. The effects of cocaine and fenfluramine but not sumatriptan were blocked in slices treated with PCA.
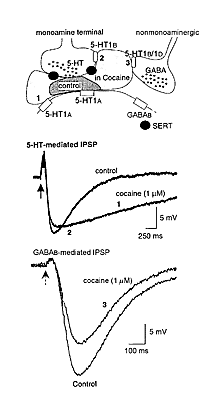 Figure 1. Cocaine dramatically changes monoamine- mediated synaptic transmission. At the top is a schematic that shows a monoamine (5-HT) terminal that releases 5-HT onto a postsynaptic cell having 5-HT1a receptors (1). The monoamine terminal has both the serotonin transport protein (SERT) and 5-HT1b/1d autoreceptors that inhibit 5-HT release (2). The schematic also shows a nonmonoamine terminal that has 5-HT1b/1d receptors that inhibit the release of GABA (3). The darker shaded area indicates the area over which 5-HT diffuses following release under control conditions. The lighter shaded area indicates that the spread of 5-HT is dramatically increased following the blockade of the SERT with cocaine. The traces in the middle are two superimposed voltage recordings of 5-HT-mediated IPSPs from a tertiary cell in the VTA. The IPSP recorded in the presence of cocaine is longer lasting (1) and smaller (2) than in control. The traces at the bottom are GABAb- mediated IPSPs recorded in a dopamine cell in the VTA. In this case cocaine decreased the amplitude of the GABAb IPSP without changing the timecourse. This effect results from a presynaptic inhibition of GABA release caused by the activation of 5-HT1b/1d receptors located on the GABA-releasing terminal (3).
Figure 1. Cocaine dramatically changes monoamine- mediated synaptic transmission. At the top is a schematic that shows a monoamine (5-HT) terminal that releases 5-HT onto a postsynaptic cell having 5-HT1a receptors (1). The monoamine terminal has both the serotonin transport protein (SERT) and 5-HT1b/1d autoreceptors that inhibit 5-HT release (2). The schematic also shows a nonmonoamine terminal that has 5-HT1b/1d receptors that inhibit the release of GABA (3). The darker shaded area indicates the area over which 5-HT diffuses following release under control conditions. The lighter shaded area indicates that the spread of 5-HT is dramatically increased following the blockade of the SERT with cocaine. The traces in the middle are two superimposed voltage recordings of 5-HT-mediated IPSPs from a tertiary cell in the VTA. The IPSP recorded in the presence of cocaine is longer lasting (1) and smaller (2) than in control. The traces at the bottom are GABAb- mediated IPSPs recorded in a dopamine cell in the VTA. In this case cocaine decreased the amplitude of the GABAb IPSP without changing the timecourse. This effect results from a presynaptic inhibition of GABA release caused by the activation of 5-HT1b/1d receptors located on the GABA-releasing terminal (3).Thus, one more effect of low concentrations of cocaine in the VTA is to inhibit GABA release. This effect of cocaine would be expected to result in disinhibition of dopamine cells to increase the release of dopamine.
Cocaine therefore has at least three actions on synaptic transmission in the VTA. Cocaine augments 5-HT-mediated IPSPs, augments autoreceptor-mediated inhibition of 5-HT release, and facilitates heterosynaptic inhibition of GABA release mediated by 5-HT1d receptors. Although not discussed here, similar mechanisms have been either demonstrated or predicted to occur at many sites in the CNS for each of the monoamine transmitters.
Adaptations Resulting From Chronic Cocaine Treatment
The mesolimbic system is known to play a role in self-administration of many drugs of abuse, including cocaine and opioids. Although morphine and cocaine act by separate cellular mechanisms initially, there must be a common pathway that results in the activation of reward pathways. One way to examine the potential common effects is to identify the common adaptations that result from repeated administration of drugs of abuse. Following chronic treatment with cocaine or morphine, a common change in synaptic regulation of dopamine cells in the VTA was observed 1 week after termination of chronic treatment. Normally D1 receptor activation augmented the amplitude of a GABAb IPSP (Cameron and Williams 1993), but in drug-experienced animals, D1 receptor activation caused an inhibition of the GABAb IPSP (Bonci and Williams 1996). The inhibition was blocked by adenosine A1-receptor antagonists and agents that disrupted the metabolism of cAMP. Thus, it appears that there is a long-lasting change in the balance between the augmentation of GABA release caused by D1-receptor activation and the inhibition mediated by adenosine that is produced by the metabolism of cAMP released from the GABA-containing nerve terminals that project from the nucleus accumbens or ventral pallidum.
This long-lasting dopamine/adenosine interaction may be one mechanism involved in dopamine-mediated craving and relapse to drug-seeking behaviors. This study suggests that neurochemical mechanisms that may be unrelated to the initial action of cocaine on the dopamine system, such as the augmentation of adenosine tone, can result in a persistent change in the synaptic regulation of dopamine cell activity.
Acknowledgment
This research was supported by National Institute on Drug Abuse Grant No. DA-04523.
References
- Bonci, A., and Williams, J.T. A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron 16:631-639, 1996.
- Cameron, D.L.; Wessendorf, M.W.; and Williams, J.T. A subset of ventral tegmental area neurons is inhibited by dopamine,
- 5-hydroxytryptamine and opioids. Neuroscience 77:155-166, 1997.
- Cameron, D.L., and Williams, J.T. Dopamine D1 receptors facilitate transmitter release. Nature 366:344-347, 1993.
- Cameron, D.L., and Williams, J.T. Cocaine inhibits GABA release in the VTA through endogenous 5-HT. J Neurosci14:6763- 6767, 1994.
- Regulation of the Dopamine Transporter and Cocaine Sensitization
-
Nancy R. Zahniser, Ph.D.
University of Colorado School of Medicine
Denver, COOur particular interest is in determining how changes in dopamine transporter (DAT) function may contribute to cocaine sensitization (Zahniser et al. 1995). The DAT is critical in determining the concentration of extracellular dopamine (DA) and, thus, overall dopaminergic tone. Cocaine blocks all three neuronal membrane monoamine transporters with approximately equal affinity. By blocking the DAT, cocaine allows released DA to persist in the extracellular space, prolongs DA receptor stimulation, and results in behavioral activation.
Repeated, intermittent administration of cocaine often results in enhanced locomotor responsiveness or behavioral sensitization (Kalivas and Stewart 1991). This phenomenon is not unique to cocaine; other psychomotor stimulants, some other classes of drugs, and stress all can induce behavioral sensitization. One of the hallmarks of behavioral sensitization is its persistence. Sensitized behavioral responses can be observed in response to a challenge dose of cocaine administered days to months after withdrawal from repeated drug treatment. However, it should be noted that, at least with the cocaine treatment and withdrawal regimens that we have used, some of the rats (25 to 50 percent) do not exhibit behavioral sensitization (Cass et al. 1993a). These "nonsensitized" rats generally demonstrate relatively more activation than the "sensitized" rats to the initial cocaine injection.
Behavioral sensitization likely reflects synaptic plasticity and, in fact, shares some properties with long-term potentiation, a more well-known type of synaptic plasticity. Many changes associated with psychomotor stimulant-induced behavioral sensitization-including neurochemical, electrophysiological, molecular, and morphological changes-have been identified. Besides being of interest to basic neuroscientists, behavioral sensitization may have clinical relevance. Several lines of evidence (Robinson and Berridge 1993) suggest that sensitization plays a role in human cocaine abuse. In particular, behavioral sensitization may contribute to the intense craving, high relapse rate, and paranoid psychosis associated with cocaine abuse.
Do Changes In DAT Function Contribute To Expression Of Cocaine Sensitization?
Since cocaine directly interacts with the DAT, we hypothesized that repeated cocaine administration might induce long-lasting changes in the activity of the DAT and that these changes might contribute to the expression of behavioral sensitization. Whereas changes at DA neuronal cell bodies are critical for initiation of sensitization, changes in the brain regions containing the DA neuronal terminals are thought to mediate the persistence, or expression, of sensitization (Kalivas and Stewart 1991). Thus, we focused on the DAT in dorsal striatum and nucleus accumbens. To address our hypothesis, Wayne Cass, in collaboration with Greg Gerhardt, developed an electrochemical method using high-speed chronoamperometry for measuring in vivo clearance of exogenously applied DA. We carried out a number of studies to demonstrate that DA clearance measured with this method reflects the activity of the DAT (Cass et al. 1993b).
To measure exogenous DA clearance, electrode-pipette assemblies were constructed, calibrated in vitro, and then implanted under stereotaxic control into dorsal striatum and core of the nucleus accumbens of urethane-anesthetized rats. The electrode-pipette assembly consisted of a glass micropipette glued a fixed distance (~300 &m) from a single, Nafion-coated carbon fiber electrochemical electrode. Pressure ejection of finite volumes of DA (200 &M barrel concentration, 12-200 nl) once every 5 minutes resulted in transient and reproducible DA signals that rose rapidly to approximately 2 &M and then decayed within 0.5 to 1.5 minutes. DA clearance rate was determined from the slope of the initial linear portion of the decay curve, that is, the slope between the points when the signal had decayed by 20 percent and 60 percent from its maximal amplitude. The density of DATs in dorsal striatum is 40 percent higher than in nucleus accumbens, and consistent with this observation, baseline clearance rates were 80 percent faster in dorsal striatum (164 Å 29 nM/s, n = 15) than in nucleus accumbens (90 Å 14 nM/s). DA clearance rates were measured in rats that received a single acute injection of either saline (1 ml/kg, i.p.) or cocaine (10 or 20 mg/kg, i.p.). The effect of the 10 mg/kg dose did not differ from that of saline in either brain region. However, clearance rate only in nucleus accumbens decreased to a greater extent in response to the 20 mg/kg dose of cocaine compared with the 10 mg/kg dose (see figure 1). Thus, the nucleus accumbens with its lower density of DATs was more sensitive to the effects of cocaine, suggesting that there may be "spare" transporters in the dorsal striatum. We have also observed a greater sensitivity of nucleus accumbens, versus dorsal striatum, with local application of cocaine (Cass et al. 1993b).
DA clearance was determined following withdrawal from repeated cocaine administration (Cass et al. 1993a). Rats received once-daily intraperitoneal injections of either saline or cocaine (10 mg/kg) for 1 week. Treatment was withheld for 1 week, and then a challenge dose of either saline or cocaine (10 mg/kg), respectively, was administered while the rats were anesthetized during the in vivo electrochemical recording session on day 15. Saline challenge, following repeated saline injection and withdrawal, did not alter DA clearance rate in nucleus accumbens. In contrast, the 10 mg/kg challenge dose of cocaine significantly attenuated clearance in the nucleus accumbens of the rats that had been withdrawn from repeated cocaine treatment (see figure 1). In fact, the 10 mg/kg cocaine challenge dose decreased DA clearance rate in the repeatedly treated/withdrawn rats to an extent similar to that of the acute 20 mg/kg dose in naive rats (figure 1). This result is consistent with cocaine-induced behavioral sensitization; diminished DA clearance would be expected to result in higher extracellular concentrations of DA and greater behavioral activation.
Conclusions and Future Directions
By analogy to our initial findings in dorsal striatum and nucleus accumbens discussed above, one explanation for a greater sensitivity of the DAT to the challenge dose of cocaine in the nucleus accumbens of rats treated repeatedly and withdrawn from cocaine would be a decreased density of DATs. Several hours after the conclusion of the in vivo electrochemical experiments, the rats were sacrificed, and quantitative autoradiography with [3H]mazindol was used to study the DATs. No differences were detected in the density of DATs or in the affinity of cocaine for the DAT in nucleus accumbens (Cass et al. 1993a). However, several other groups have observed decreased densities of DATs selectively in nucleus accumbens following 10 to 60 days of withdrawal from repeated cocaine administration (Kuhar and Pilotte 1996). Furthermore, this is one of the most persistent neurochemical alterations, with a duration similar to that of behavioral sensitization, that has been identified following repeated cocaine administration (Kuhar and Pilotte 1996).
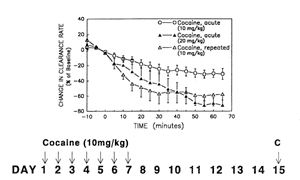 Figure 1. Enhanced inhibition of DA clearance in nucleus accumbens of rats given a challenge dose of cocaine following withdrawal from repeated cocaine treatment compared with acute administration of cocaine. Groups of rats were injected i.p. with the dose of cocaine indicated. The repeated cocaine treatment regimen is indicated below the graph; the challenge dose (C) was administered on day 15. Mean values ± SEM are shown for n = 4-5.
Figure 1. Enhanced inhibition of DA clearance in nucleus accumbens of rats given a challenge dose of cocaine following withdrawal from repeated cocaine treatment compared with acute administration of cocaine. Groups of rats were injected i.p. with the dose of cocaine indicated. The repeated cocaine treatment regimen is indicated below the graph; the challenge dose (C) was administered on day 15. Mean values ± SEM are shown for n = 4-5.Greater cocaine-induced inhibition of DA clearance was observed in the nucleus accumbens of all rats withdrawn from repeated cocaine treatment when compared with either administration of saline or acute administration of the same dose of cocaine. This result is consistent with cocaine-induced behavioral sensitization. However, since the rats were anesthetized during the electrochemical experiment when the challenge dose of cocaine was administered, it was impossible to determine whether these rats exhibited behavioral sensitization in response to the challenge dose. Their behavior had been monitored on the first and last days of the repeated treatment, and only about half of the rats exhibited behavioral sensitization (Cass et al. 1993a). Therefore, to test our hypothesis further we need to measure DA clearance and behavior concomitantly. Greg Gerhardt, Charlie Ksir, and Chad Pivik have developed the technology to measure DA clearance in the freely behaving rat. The electrode-pipette assembly is fabricated from fused silica tubing, and the headstage has been miniaturized. An acute injection of cocaine induces a simultaneous change in both locomotor activity (measured with a computer-based activity chamber) and exogenous DA clearance in nucleus accumbens. The altered clearance appears to be somewhat longer lasting than the altered behavior, as observed by other groups using in vivo microdialysis to measure extracellular endogenous DA concentrations in freely behaving rats. Shelly Dickinson has recently established this methodology in my laboratory and is currently conducting experiments to test the long-term stability of the in vivo exogenous DA clearance measurements in the chronically implanted rats. This approach can be used to determine whether the cocaine-induced alterations in DAT function are associated with expression of behavioral sensitization.
By What Mechanisms Are DAT Activity And Expression Regulated And Modulated?
The idea that regulation of DAT activity and expression might contribute to cocaine-induced behavioral sensitization leads to curiosity about the mechanisms that might be involved generally in DAT regulation. In 1993 I collaborated with Susan Amara, Mark Sonders, and Mike Kavanaugh at the Vollum Institute, Oregon Health Sciences University, to show that the human DAT (hDAT) expressed in Xenopus laevis oocytes is electrogenic (Sonders et al. 1997). As expected based on other investigators' conclusion that DAT cotransports two Na+ ions, one Clå ion, and one DA+ molecule, we found (using a two-electrode voltage clamp) small, but measurable and concentration- dependent, inward currents induced by DA and other DAT substrates. The transport-associated currents were voltage dependent, increasing at more hyperpolarized potentials. Somewhat more unexpected, however, was the observation that DAT blockers, like cocaine, induced small outward currents in hDAT-expressing oocytes. This was shown to be due to blockade of an inward cation leak associated with hDAT expression. Blockade of this leak current by substrates was also apparent at more depolarized holding potentials (>0 mV) and under conditions when transport was abolished, such as substitution of Na+ by Li+ in the buffer. An important prediction from these observations was that DA uptake by the hDAT should also be voltage dependent. Si-Jia Zhu, working in my laboratory, measured uptake in the hDAT-expressing oocytes under voltage clamp and found that this was indeed the case. Hyperpolarization increased the velocity of the transporter, whereas depolarization decreased velocity (Sonders et al. 1997). A change of 30 mV altered DA uptake by 25 percent. These results suggest one mechanism, changes in membrane potential, that may transiently regulate the activity of the DAT.
In order to determine whether activity of the DAT in the brain is also regulated by membrane potential, Alex Hoffman, Greg Gerhardt, Carl Lupica, and I have begun to carry out electrochemical measurements in rat brain slices containing the substantia nigra pars compacta (SNc) (Zahniser et al. 1998). Characterization experiments indicate that the DAT is still the major determinant of exogenous DA clearance in the SNc. The patch-clamp method was used to monitor changes in the membrane potential of SNc neurons in the hemisphere opposite to that where DA clearance was measured. Neurons were identified as being DA-like by their characteristically long-duration action potentials. As our first approach to alter membrane potential, the effect of increasing the concentration of KCl in the superfusion buffer was investigated. Tetrodotoxin (0.5 &M) was included throughout these experiments to block voltage-gated sodium channels and to ensure that any effects of KCl on DA clearance were due to direct voltage changes and not to changes in Na+ gradients. Elevating the KCl concentration to 30 mM resulted in a 30-mV depolarization and decreased DA clearance by 35 percent. Although preliminary, these results suggest that DAT velocity in the rat brain, similar to that of the hDAT expressed in oocytes, is transiently reduced by membrane depolarization. Future experiments will use different strategies to hyperpolarize, as well as to depolarize, membrane potential. The idea that the DAT is voltage dependent is also interesting because it provides a potential mechanism by which presynaptic receptors, such as D2 DA autoreceptors, could influence DAT activity. Several groups have demonstrated that D2 receptor ligands can modulate rat striatal DA clearance measured both in vitro and in vivo (Cass and Gerhardt 1994). Their results demonstrate that activation of D2 receptors decreases, whereas antagonism of these receptors increases, DA clearance rate. This is exactly what we would have predicted based on the fact that D2 receptors open potassium channels, which hyperpolarize the membrane. Shelly Dickinson has employed another approach to address the question of D2 DA receptor/DAT interactions. She has measured in vivo DA clearance in dorsal striatum of the D2 receptor knockout mice produced by Malcolm Low, David Grandy, Michele Kelly, and Marcelo Rubinstein of the Vollum Institute. She found that a significantly larger volume of DA had to be ejected in the wildtype than in the knockout mice to produce a maximal signal amplitude of 2-3&M. The DA clearance rate was also higher in the wildtype mice. These results are consistent with the idea that the exogenous DA that we normally apply to measure clearance is interacting with D2 receptors, accelerating DAT velocity, and thereby increasing DA clearance. In the knockout mice, DAT clearance is attenuated because they lack D2 receptors. Thus, our results again support the idea that release of DA and activation of D2 autoreceptors produce two effects that would reduce extracellular concentrations of DA-inhibition of further DA release and acceleration of DA uptake. Dayne Mayfield is currently investigating the effects of coexpressing the D2 DA receptor and appropriate potassium channels, along with the hDAT, in oocytes, since in this system the contribution of changes in membrane potential to D2 receptor/DAT interactions can be evaluated.
Phosphorylation of the DAT is perhaps the most obvious mechanism for regulation of its activity. The hDAT contains two consensus sites for phosphorylation by protein kinase C (PKC). Several groups-using rat striatal synaptosomes, primary mesencephalic cultures, or heterologous cell expression systems-have shown that DAT activity is sensitive to PKC activation (Zhu et al. 1997). Si-Jia Zhu used the oocyte expression system to investigate the mechanism by which PKC regulates the hDAT (Zhu et al. 1997). She found that bath application of the PKC activator phorbol 12-myristate 13-acetate (PMA) dose and time dependently diminished [3H]DA uptake in the hDAT-expressing oocytes. The IC50 was 22 nM, and 100 nM PMA produced a maximal effect. The effect of PMA (100 nM) appeared to be due to activation of PKC because it was partially reversed by the selective PKC inhibitor bisindolylmaleimide (1&M) and was not mimicked by the inactive PMA analog 4-ä-phorbol-12,13-didecanoate (400 nM). The reduction in [3H]DA uptake was due to decreased maximal velocity of transport (Vmax), with no change in transporter affinity. Exposure to 100 nM PMA for 30 minutes reduced Vmax by 69 Å 11 percent. Similarly, exposure to 100 nM PMA for 3 to 5 minutes, followed by 15 to 30 minutes of washing, reduced both the transport- associated and leak currents by 50 to 80 percent. Changes in oocyte cell surface area induced by PMA exposure were determined from capacitive transients. Baseline membrane capacitance did not differ among uninjected oocytes, hDAT-expressing oocytes, uninjected oocytes exposed to PMA, and hDAT-expressing oocytes exposed to PMA. Pretreatment for 3 to 5 minutes with 100 nM PMA produced a time-dependent decrease in membrane capacitance selectively in the hDAT-expressing oocytes; after 30 minutes of washing, the loss was approximately 40 percent. Saturation curves generated with [3H]mazindol in intact oocytes revealed a 78-percent decrease in the density of binding sites, with no change in affinity, following PMA pretreatment. However, no change in [3H]mazindol binding was observed in homogenates, which would contain both cell surface and internal membranes, from oocytes exposed to PMA. Taken together, these results suggest that PMA, via activation of PKC, alters cell surface trafficking of the hDAT, rather than converting the hDAT to a less active state.
Conclusions and Future Directions
Our studies suggest several mechanisms by which DAT activity can be regulated. DAT velocity is transiently altered by changes in membrane potential and by activation of D2 DA receptors. Whether these two observations are related will require additional experimentation in the hDAT-expressing oocytes and rat brain slices. However, our results demonstrate that depolarization diminishes DAT activity, whereas hyperpolarization accelerates it. Our results also support the notion that increases in extracellular DA could produce two effects via activation of D2 autoreceptors, inhibition of further DA release and acceleration of DA uptake, that would work in concert to reduce extracellular DA and limit postsynaptic receptor activation.
Activation of PKC reduces hDAT activity in the oocyte expression system by inducing changes in membrane trafficking of the transporter. These observations raise the possibility that PKC-coupled presynaptic receptors, such as Class 1 metabotropic glutamate receptors, could mediate decreases in DAT expression that, in turn, would be expected to enhance and prolong DA neurotransmission. PKC-induced increases in endocytosis have been observed with three other Na+/Clå-dependent neurotransmitter transporters-GABA, taurine, and serotonin transporters-and the Na+/glucose cotransporter (Zhu et al. 1997). All of this evidence suggests that alterations in membrane trafficking of transporters is an effect common to PKC activation. Whether this mechanism, or another similar phosphorylation-dependent mechanism, mediates the persistent changes in DAT activity and/or expression induced by repeated cocaine administration remains to be explored in future experiments.
Acknowledgments
Many wonderful collaborators have participated in this work. They include Wayne Cass, Greg Gerhardt, Kelly Gillespie, Pam Curella, Dayne Mayfield, Karen Flach, Gaynor Larson, Si-Jia Zhu, Shelly Dickinson, Jilla Sabeti, Alex Hoffman, and Carl Lupica at the University of Colorado; Charlie Ksir and Chad Pivik at the University of Wyoming; and Susan Amara, Mark Sonders, Mike Kavanaugh, Malcolm Low, David Grandy, Michele Kelly, and Marcelo Rubinstein at the Vollum Institute, Oregon Health Sciences University. The work from my lab was supported by National Institutes of Health Grant No. DA-04216, career development award DA-00174, and postdoctoral fellowship DA-05706.
References
- Cass, W.A., and Gerhardt, G.A. Direct in vivo evidence that D2 dopamine receptors can modulate dopamine uptake. Neurosci Lett 176:259-263, 1994.
- Cass, W.A.; Gerhardt, G.A.; Gillespie, K.; Curella, P.; Mayfield, R.D.; and Zahniser, N.R. Reduced clearance of exogenous dopamine in rat nucleus accumbens, but not in dorsal striatum, following cocaine challenge in rats withdrawn from repeated cocaine administration. J Neurochem 61:273-283, 1993a.
- Cass, W.A.; Zahniser, N.R.; Flach, K.A.; and Gerhardt, G.A. Clearance of exogenous dopamine in rat dorsal striatum and nucleus accumbens: Role of metabolism and effects of locally applied uptake inhibitors. J Neurochem 61:2269-2278, 1993b.
- Kalivas, P.W., and Stewart, J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Brain Res Rev 16:223-244, 1991.
- Kuhar, M.J., and Pilotte, N.S. Neurochemical changes in cocaine withdrawal. Trends Pharmacol Sci 17:260-264, 1996.
- Robinson, T.E., and Berridge, K.C. The neural basis of drug craving: An incentive-sensitization theory of addiction. Brain Res Brain Res Rev 18:247-291, 1993.
- Sonders, M.S.; Zhu, S.-J.; Zahniser, N.R.; Kavanaugh, M.P.; and Amara, S.G. Multiple ionic conductances of the human dopamine transporter: The actions of dopamine and psychostimulants. J Neurosci 17:960-974, 1997.
- Zahniser, N.R.; Gerhardt, G.A.; and Cass, W.A. Chronic cocaine action on the dopamine transporter. In: The Neurobiology of Cocaine: Cellular and Molecular Mechanisms. Hammer, Jr., R.L., ed. Boca Raton, FL: CRC Press, 1995. pp. 181-197.
- Zahniser, N.R.; Gerhardt, G.A.; Hoffman, A.F.; and Lupica, C.R. Voltage-dependency of the dopamine transporter in rat brain. In: Advances in Pharmacology, Vol. 42. Goldstein, D.; Eisenhofer, G.; and McCarty, R., eds. San Diego, CA: Academic Press, 1998. pp. 195-198.
- Zhu, S.-J.; Kavanaugh, M.P.; Sonders, M.S.; Amara, S.G.; and Zahniser, N.R. Activation of protein kinase C inhibits uptake, currents and binding associated with the human dopamine transporter expressed in Xenopus oocytes. J Pharmacol Exp Ther 282:1358-1365, 1997.
- Whole-Cell Plasticity in Cocaine Addiction: Current Perspectives
-
Francis J. White, Ph.D.
Finch University of Health Sciences
The Chicago Medical School
North Chicago, ILIntroduction
Cocaine addiction remains one of the foremost public health problems in the United States. Cocaine dependence is typically associated with cyclical patterns of drug use and abstinence. During abstinence, periods of intense cocaine craving and other withdrawal symptoms (anergia, anhedonia, and depression) often lead to relapse. Animal models of cocaine dependence have successfully identified several neuroadaptations in brain circuitry involved in cocaine addiction, particularly within the dopamine (DA) neuronal system projecting from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) and related limbic and cortical areas. These neuroadaptations range from changes in transporters, receptors, and transduction molecules to alterations in gene expression. Despite our increasing knowledge, pharmacotherapies targeted at such changes have yet to prove effective in cocaine-dependent individuals, in part because of our incomplete understanding of the relevant alterations and how they occur.
We have used electrophysiological techniques to study alterations in the mesoaccumbens DA system at the level of single neurons, particularly within the NAc. We have focused on the NAc because of its known role in cocaine self-administration and other forms of motivated behavior. NAc neurons receive convergent excitatory commands from the ventral hippocampus (subiculum), basolateral amygdala, and prefrontal cortex, which together supply information regarding environmental conditions, behavioral contingencies, emotional states, and motivational (homeostatic) needs. DA from the VTA neurons modulates such information by using a rich repertoire of presynaptic and postsynaptic mechanisms.
Nucleus Accumbens Neurons
A great deal of information regarding NAc neurons has emerged over the past half decade. The major neuronal population in the NAc is the medium spiny GABAergic output neuron. These neurons encode excitatory commands and relay information to two nuclei of the basal ganglia, the ventral (subcommisural) pallidum and the substantia nigra pars reticulata, as well as to the medial-dorsal thalamic nucleus, creating a loop back to the cortex. Like their neighbors in the caudate nucleus, medium spiny neurons of the NAc exhibit a two-state spontaneous membrane potential, fluctuating between a highly hyperpolarized state (-75 to -90 mV, the down-state) and a more depolarized state (-45 to -60 mV, the up-state) close to the firing threshold. The down-state represents periods of relatively little excitatory input and is maintained by an inwardly rectifying K+ conductance. Transition to the up-state and to spike activation requires a convergent barrage of maintained excitation from hippocampus, amygdala, and/or prefrontal cortex, with hippocampal information playing a strategic "gating" role for spike activity (O'Donnell and Grace 1995). However, maintenance of the up-state membrane potential requires interplay between several voltage-dependent inward and outward currents, which are subject to neuromodulation. Given the importance of such conductances in controlling the state of activity of NAc output neurons, we decided to pursue a program of study designed to characterize the modulation of various voltage-dependent channels, particularly by DA receptors, and to determine whether the channels and their modulation are altered by repeated cocaine administration.
Studies in Brain Slices
Our first experiments were conducted in brain slices. In this in vitro preparation, medium spiny NAc neurons exist only in the down-state because most excitatory afferents are severed. We used traditional current-clamp intracellular recordings to compare the membrane properties of NAc neurons in rats that received once-daily cocaine (15 mg/kg) or saline (control) injections for 5 days. All experiments were conducted on the third day after the last injection of cocaine. We found that the resting membrane potentials of cocaine-withdrawn NAc neurons were more hyperpolarized compared with control neurons. When we injected depolarizing current into the neurons to switch them to the up-state and induce action potentials (Na+ spikes), we found that cocaine- withdrawn neurons required significantly greater currents to evoke spikes. In addition, the threshold for producing action potentials was significantly increased, and the spikes were of smaller amplitude (see table 1). With sustained depolarization, control neurons exhibited repetitive firing, with the number of spikes increasing as a function of the intensity of current applied. Repetitive firing was either completely absent or markedly reduced in cocaine-withdrawn neurons. When voltage-sensitive sodium channels (VSSCs) were blocked by tetrodotoxin (TTX), we found that calcium plateau potentials were also reduced in both amplitude and duration. None of these alterations was observed in rats that received repeated administration of lidocaine, indicating that they were not the result of local anesthetic actions of cocaine.
TABLE 1. Repeated Cocaine Administration In Vivo Alters Membrane Properties of NAc Neurons Recorded In Vitro.
Measures Saline Cocaine Lidocaine Number of neurons 36 29 14 Number of rats 25 19 4 RMP (mV) -79.1 ± 0.8 -83.8 ± 0.6** -76.9 ± 1.5 Current to generate AP (nA) 0.65 ± 0.03 0.96 ± 0.06** 0.69 ± 0.09 AP threshold (mV) -46.7 ± 1.5 -40.9 ± 1.7* -46.1 ± 2.1 AP amplitude (mV) 61.1 ± 1.1 52.5 ± 1.7** 59.2 ± 1.2 NOTE: Values represent the mean ± SEM for the number of neurons indicated (* p < 0.05, ** p less than 0.01 with Student's t-test used to compare with control). Resting membrane potential (RMP) was measured in the absence of injected current prior to the initiation of other manipulations. Action potentials (APs) were generated by injecting step depolarizing current pulses of 0.1 nA increments (adapted from Zhang et al. 1998).
Studies In Dissociated Neurons
The results of our brain slice experiments clearly indicated a reduction in the excitability of NAc neurons from cocaine-withdrawn rats and suggested alterations in voltage-sensitive channels. Therefore, we initiated a series of studies to evaluate whether various channels are altered by repeated cocaine administration. To date, we have characterized VSSCs and voltage-sensitive calcium channels (VSCCs). These experiments were conducted in acutely dissociated neurons using the whole-cell configuration of the patch-clamp technique and specific agents to block all but the channels of interest. This preparation afforded us considerable control in voltage-clamp mode without concerns of poor space-clamp and allowed us to conduct mechanistic studies of neuromodulation because we had access to the internal milieu of the neurons via the patch pipette.
We first demonstrated that, as in the caudate nucleus (Surmeier and Kitai 1994), DA D1 receptors reduce whole-cell Na+ current in medium spiny neurons of the NAc. The reduction occurs because of activation of the adenylyl cyclase-cAMP transduction pathway causing enhanced phosphorylation of VSSCs by cAMP-dependent protein kinase (PKA). In cocaine-withdrawn neurons, we observed a 37-percent reduction in Na+ current density and a depolarizing shift (of about 5 mV) in the voltage dependence of activation, with no alteration in the voltage dependence of inactivation (see figure 1). This profile of effects on VSSCs has been suggested by some investigators to indicate enhanced phosphorylation by protein kinase C (PKC). However, we found that activation of PKC by phorbol esters reduced Na+ current and delayed inactivation but did not alter the voltage dependence of activation. Thus, we interpret the reduction in basal Na+ current produced by repeated cocaine administration as an indication of enhanced basal state of phosphorylation by PKA because the cAMP-PKA pathway in the NAc is clearly upregulated by repeated cocaine treatment, with increases in levels of both cAMP and PKA (Terwilliger et al. 1991). Additional study will be required to elucidate the mechanism underlying the depolarizing shift in the voltage dependence of activation. Whatever the mechanism, this result is consistent with those obtained in brain slices (above), as well as those observed in vivo (White et al. 1995), all of which indicate a reduced excitability of NAc neurons during cocaine withdrawal (Zhang et al. 1998).
Our next series of experiments examined the effects of repeated cocaine administration on isolated VSCCs. Studies of modulation of VSCCs in medium spiny neurons of the NAc yielded results that again were highly similar to those previously reported for the caudate nucleus (Surmeier and Kitai 1994). DA D1 receptors suppressed whole-cell Ca2+ currents, particularly those carried by N and P/Q type channels. This modulation occurred via cAMP-PKA-protein phosphatase cascade leading to dephosphorylation of the channels and a reduction in current. After repeated cocaine administration, the basal level of whole-cell Ca2+ current was significantly reduced, and the N-type channel appeared to be most affected. Because these channels are primarily involved in transmitter release, a reduction in current would result in reduced GABA release from these neurons when they are active.
Conclusions
Our studies have demonstrated a novel form of whole-cell (nonsynaptic) plasticity produced by repeated cocaine administration reduced Na+ and Ca2+ currents in NAc neurons. This nonsynaptic plasticity should be considered an important contributor to behavioral manifestations of cocaine addiction and withdrawal. Consider that, while synaptic plasticity will modify the responsiveness of NAc neurons to selected inputs at specific synapses, the reduction of whole-cell Na2+ currents will produce an indiscriminate decrease in the responsiveness of the NAc to excitatory commands because these channels govern the initiation of action potentials. Even when activated, the neurons are likely to transmit less information to their targets because of the reduction of GABA release due to reduced Ca2+ currents. In addition, reduced Na+ and Ca2+ conductance could also impact synaptic plasticity (such as long-term potentiation or depression), because VSSCs expressed within neuronal soma and dendrites carry backpropagating action potentials that modulate dendritic Ca2+ influx. Thus, even if whole-cell plasticity does not persist with longer withdrawal, it will help to determine which synapses undergo long-term associative synaptic plasticity as well as the magnitude of such changes.
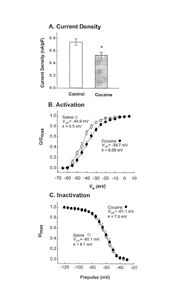 Figure 1. Repeated cocaine treatment reduced NAc Na+ currents and caused a depolarizing shift in the voltage dependence of activation
Figure 1. Repeated cocaine treatment reduced NAc Na+ currents and caused a depolarizing shift in the voltage dependence of activation
A. NAc neurons(N=33) from cocaine-pretreated rats exhibited significantly (t68 = 2.75, p= 0.0075) reduced peak whole-cell Na+ currents (measured at -20 mV) compared with neurons (N=37) obtained from saline-pretreated rats.
B. Cocaine pretreatment caused a depolarizing shift in the voltage dependence of activation and reduced the slope factor (k). Values were obtained for each neuron at each membrane voltage, and the means SEM were plotted. Half maximal activation (V1/2) and slope (k) values were obtained individually for each neuron and were compared with t-tests. Half-maximal activation occurred at significantly (t28=2.26, p=0.031) more depolarized potentials in cocaine-pretreated neurons (-40.3 1.8mV, N=16) compared with saline-pretreated neurons (46.1 1.2, N=14). The slope factor was also significantly decreased (t28=3.13, p=0.004) in the cocaine group (8.85 0.8) compared with the saline group (4.01 0.8). Note that the V1/2 and k values shown in the figure were derived from the curves fitted to the mean values depicted in the figure, not from the mean values (given here) obtained by averaging all of the neurons in the sample.
C. The voltage dependence of inactivation was not altered by repeated cocaine treatment. Values were obtained from the means SEM as in B (N = 16 for cocaine and 13 for saline). Note that repeated administration of lidocaine failed to produce effects similar to those of cocaine (adapted from Zhang et al. 1998).Given that the NAc is a structure in which functionally distinct ensembles of neurons are recruited by convergent excitatory inputs to coordinate patterns of movement and affect, a reduction in the excitability of NAc neurons would decrease the processing of such information and thereby lead to cocaine withdrawal effects such as anergia, anhedonia, and depression. In rat models, such deficits are observed during the early days of withdrawal from repeated cocaine administration when Na+ and Ca2+ currents are depressed. Moreover, given that D1 receptor stimulation further reduces both Na+ and Ca2+ currents in the cocaine-withdrawn NAc neurons, we believe that the proposed use of such drugs as a replacement therapy for cocaine-dependent individuals may be contraindicated, at least during the early period of cocaine withdrawal.
Acknowledgments
This research was supported by National Institute on Drug Abuse Grant No. DA-04093 and Research Scientist Development Award DA-00207.
References
- O'Donnell, P., and Grace, A.A. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: Hippocampal gating of prefrontal cortical input. J Neurosci 15:3622-3639, 1995.
- Surmeier, D.J., and Kitai, S.T. Dopaminergic regulation of striatal efferent pathways. Curr Opin Neurobiol 4:915-919, 1994.
- Terwilliger, R.; Beitner-Johnson, D.; Sevarino, K.A.; Crain, S.M.; and Nestler, E.J. A general role for adaptations in G-proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function. Brain Res 548:100-110, 1991.
- White, F.J.; Hu, X-T.; Zhang, X-F.; and Wolf, M.E. Repeated administration of cocaine or amphetamine alters neuronal responses to glutamate in the mesoaccumbens dopamine system. J Pharmacol Exp Ther 273:445-454, 1995.
- Zhang, X-F.; Hu, X-T.; and White, F.J. Whole-cell plasticity in cocaine withdrawal: Reduced sodium currents in nucleus accumbens neurons. J Neurosci 18:488-498, 1998.
- Cocaine Dependence and Withdrawal: Neuroadaptive Changes in Brain Reward and Stress Systems
-
Friedbert Weiss, Ph.D.
The Scripps Research Institute
La Jolla, CAA growing body of evidence indicates that chronic cocaine administration can produce profound and long-lasting changes in brain neurochemical and neuroendocrine systems. At the behavioral level, evidence is accumulating that chronic use of cocaine compromises the neural mechanisms that mediate positive reinforcement. This is illustrated, for example, by findings that cocaine acutely facilitates the rewarding effects of intracranial self-stimulation, while withdrawal after chronic use leads to an impairment in the rewarding efficacy of electrical brain stimulation (Markou and Koob 1991). Findings such as these have given rise to the view that compulsive drug-seeking behavior associated with cocaine (and other drugs of abuse) may be the result of adaptive processes within the central nervous system that oppose the acute reinforcing actions of drugs, leading both to a "blunting" of mechanisms that mediate positive reinforcement and the emergence of affective changes during withdrawal that may motivate continued use of the drug (for example, anxiety, dysphoria, and depression) during withdrawal (Koob and Bloom 1988; Koob et al. 1993; Wise 1996).
The following discussion reviews both earlier and recent studies that have sought to identify the brain neurochemical processes responsible for the compromised state of the reward system after chronic cocaine abuse and the significance of those processes in the transition from controlled drug use to compulsive drug-taking.
Neuroadaptive Changes Within Brain Reward Circuitries
Intravenous self-administration in rodents has been used successfully to study cocaine-reinforced behavior. This methodology has significantly advanced the understanding of the neurobiological basis of cocaine reinforcement and has established a critical role for the mesoaccumbens dopamine (DA) systems in cocaine's acute reinforcing effects. More recently, studies employing intracranial microdialysis measures of DA in the nucleus accumbens (NAc) of cocaine self-administering rats have confirmed the significance of DA in cocaine reward and extended our understanding of interactions among cocaine, DA, and other transmitters in this brain region in the regulation of cocaine-seeking behavior.
When given the opportunity, both human cocaine abusers and laboratory animals will often self-administer cocaine in sustained episodes that can last from several hours to days. In humans, in particular, this so-called binge pattern of cocaine abuse is associated with severe abstinence syndrome. In animals, termination of access to cocaine after long-term unrestricted intravenous self-administration produces behavioral disruptions and reward deficits believed to be indicative of dependence and withdrawal (Markou and Koob 1991). Therefore, this model was employed in conjunction with intracranial microdialysis to study the neurochemical consequences of long-term cocaine self-administration and cocaine withdrawal.
Dopamine
Cocaine self-administration produced persistent elevations in extracellular DA concentrations in the NAc that remained stable throughout 12- to 24-hour periods of drug availability. Withdrawal from cocaine resulted in a marked suppression of DA release below basal levels prior to self-administration (Weiss et al. 1992b). Maximal inhibition of DA efflux was reached within 2 to 4 hours postcocaine, and the depression in extracellular DA levels did not recover within a 12-hour monitoring period. The degree of suppression of DA release was positively correlated with the number of hours of continuous cocaine self-administration before withdrawal. Interestingly, as shown in earlier work, deficits in brain stimulation reward also increased as a function of the duration of continuous self-administration prior to withdrawal and were reversible by administration of bromocriptine (Markou and Koob 1991, 1992; Weiss et al. 1995). These data implicate a link between the withdrawal-associated impairment in mesolimbic DA neurotransmission and behavioral abstinence symptoms as measured by attenuated brain stimulation reward. However, it is important to note that brain stimulation reward deficits are already evident shortly after termination of access to cocaine, at a time when there is still some residual elevation, rather than a deficit, in accumbal extracellular DA levels. This observation supports the hypothesis that sustained dopaminergic stimulation by long-term cocaine self-administration leads to adaptation of brain mechanisms that mediate positive reinforcement.
Serotonin
Cocaine self-administration not only increases extracellular DA levels in the NAc but also produces similar elevations in extracellular serotonin (5-HT). Given the established role of 5-HT in depression, a prominent cocaine withdrawal symptom, it was of interest to determine whether cocaine withdrawal exerts disruptive effects on 5-HT neurotransmission.
Withdrawal after 12 hours of unrestricted access to cocaine produced a substantial suppression of 5-HT release in the NAc. Compared with basal 5-HT levels in cocaine-naive controls, 5-HT efflux as measured by quantitative microdialysis methods decreased by more than 50 percent as early as 6 hours postcocaine (Parsons et al. 1995). In contrast to the serotonergic deficits after long-term cocaine self-administration, only a trend toward suppression of basal 5-HT release was apparent in rats after 24 hours of abstinence from daily 3-hour limited-access self-administration. These findings are consistent with several reports in the literature of supersensitivity of 5-HT1a autoreceptors and increased density of 5-HT uptake sites after intermittent cocaine administration, but they also suggest that marked extracellular consequences of these presynaptic changes become evident only after prolonged periods of continuous cocaine self-administration. Decreased serotonergic transmission has been implicated in symptoms of numerous psychiatric disorders such as depression, panic disorder, insomnia, impulsiveness, and aggression-symptoms also associated with cocaine abstinence. Therefore, the deficit in extracellular 5-HT concentrations may contribute directly to many aspects of the cocaine withdrawal syndrome.
In addition to suppressing the release of 5-HT, withdrawal after long-term access to intravenous cocaine altered the sensitivity of 5-HT1b receptors. Locomotor activation in response to a 5-HT1b agonist (RU 24969) was diminished during the first 2 days of cocaine withdrawal, while a persistent rebound supersensitivity to 5-HT1b receptor activation emerged 1 week after cocaine withdrawal. The initial subsensitivity is likely to reflect an adaptive "downregulation" of 5-HT1b receptors that develops during long-term cocaine self-administration to compensate for the sustained cocaine-induced increases in synaptic 5-HT levels. Conversely, the subsequent supersensitivity is presumably the result of sustained extracellular 5-HT deficiency during cocaine withdrawal. These findings implicate 5-HT1b receptors, both in the cocaine withdrawal syndrome and in locomotor sensitization produced by repeated cocaine administration.
Recent studies have implicated the 5-HT1b receptor in the acute reinforcing actions of cocaine. The 5-HT1b agonists produced a dose-dependent shift to the left in the dose-effect function for self-administered cocaine and elevated breaking points for cocaine on a progressive ratio schedule (Parsons et al., submitted). The enhancement of cocaine reward by 5-HT1b receptor activation appeared to result from an augmentation in the accumulation of extracellular DA in the NAc induced by cocaine, a finding that suggests that 5-HT1b receptors, via stimulation by endogenous 5-HT, may have a role in cocaine reinforcement. The subsensitivity of 5-HT1b receptors during the early withdrawal phase is, therefore, interesting, not only with regard to its role in cocaine withdrawal but also with regard to the general hypothesis that dependence may result from adaptation of central reward mechanisms.
Changes In Brain Stress Systems After Chronic Cocaine
Recently, much attention has been directed at understanding the role of the nonneuroendocrine corticotropin-releasing factor (CRF) system in the central nucleus of the amygdala (CeA) in the affective consequences of stress and in withdrawal from drugs of abuse. The CeA is part of a complex neural circuitry regulating behavioral and autonomic responsiveness to stressful stimuli. In particular, CRF neurons in the CeA are thought to have an essential role in the mediation of emotional responses to stress, such as anxiety. Anxiety and stress-like symptoms are an integral part of drug withdrawal syndromes, raising the possibility that these withdrawal signs may involve activation of CRF neuronal mechanisms in the CeA.
Initial findings indicated that acute intraperitoneal injections of cocaine increase CRF release in the CeA of rats. This effect was significantly enhanced by 2 weeks of daily cocaine pretreatment, implicating CRF mechanisms in the CeA in cocaine sensitization as well as in the cross-sensitization between stress and psychostimulants (Richter et al. 1995). In contrast to the effects of noncontingent, intermittent cocaine administration, however, CRF release in the CeA was significantly suppressed by cocaine in self-administering rats as measured after completion of 2 weeks of cocaine self-administration training. Moreover, in these animals, cocaine withdrawal after 12 hours of continuous access to the drug produced a profound increase in CRF release, which reached peak levels of approximately 400 percent of baseline between 11 and 12 hours postcocaine (Richter and Weiss, submitted).
These data provide support for involvement of CRF mechanisms in the CeA in the motivational effects of cocaine. Central administration of CRF has stress-like anxiogenic and activational consequences in rats that can be effectively reversed by treatments that interfere with CRF transmission in the CeA. The effects of exogenous CRF resemble the behavioral signs of cocaine withdrawal in animals; these effects may be comparable to human withdrawal symptoms such as anxiety, agitation, irritability, restlessness, and confusion. Thus, the activation of CRF release in the CeA during withdrawal may provide a neurochemical basis for aspects of the cocaine abstinence syndrome. In contrast, the suppression of CRF release by cocaine during the self-administration stage may implicate attenuation of CRF release in the CeA as an element in the reinforcing actions of cocaine. Finally, these data extend previous observations on the activation of CRF mechanisms in the CeA during opiate, ethanol, and cannabinoid withdrawal and implicate enhanced amygdaloid CRF release as a common mechanism in symptoms of anxiety and negative affect that are typically associated with drug withdrawal syndromes (de Fonseca et al. 1997; Merlo Pich et al. 1995).
The evidence of a hyperactivity within an important brain stress regulatory center during cocaine withdrawal is intriguing in view of the established role of stress in drug abuse and dependence. Stress is a major determinant of relapse in humans and can increase the intake of psychostimulant drugs; it can also facilitate the acquisition of psychostimulant self-administration in laboratory animals. While many stress-associated drug-seeking behaviors may involve activation of the hypothalamic CRF system and the hypothalamic-pituitary-adrenal axis, the present data support an essential role for amygdalar CRF neurons in drug-seeking behavior motivated by stress or anxiety effects related to cocaine abstinence.
Studies examining the interaction between stress and psychostimulant withdrawal indicate that, in addition to disturbances in the brain CRF system, chronic psychostimulant exposure can disrupt normal stress responses at other levels. For example, not only did termination of daily amphetamine treatment result in a long-lasting deficit in extracellular DA concentrations in the NAc, but also stimulation of DA release in response to restraint stress, which is a typical response to this stressor in drug-naive animals, was no longer observed during amphetamine withdrawal. In fact, restraint stress produced a persistent reduction in extracellular DA concentration below basal levels that were already lowered by withdrawal from chronic amphetamine (Weiss et al., in press). Thus, certain forms of stress may exacerbate the neurochemical consequences of psychostimulant withdrawal by further lowering extracellular DA levels and, thereby, perhaps contribute to the resumption of drug-seeking behavior and increased likelihood of relapse associated with stress. Moreover, the reversal of the dopaminergic response to immobilization stress was not confined to acute abstinence but was still observed at the same magnitude 7 days postamphetamine. This persistent suppression in DA release after stress may reflect a disruption of mechanisms that regulate affective homeostasis, leading to an impairment in the ability to cope with stress or emotional challenges. Such defects may have important implications for emotional states such as depression or helplessness and for vulnerability to relapse over a prolonged abstinence period.
Chronic Cocaine And Behavioral Plasticity
The data discussed above identify perturbations in brain reward and stress systems as an important element in neuroadaptive changes induced by chronic cocaine. Another important factor associated with chronic use of cocaine (and other drugs of abuse) may involve plasticity within brain circuitries that mediate conditioning effects or stimulus-response associations. Indeed, the classical conditioning of cocaine's pharmacological effects with specific drug-associated environmental stimuli is an important aspect of its behavioral actions. Cocaine-associated stimuli can mimic the drug's locomotor-activating effects and control place preference induced by repeated pairing of cocaine injections with a specific environment. The conditioning of cocaine's rewarding actions with environmental stimuli has important implications for its abuse potential. Clinical observations suggest that stimuli previously associated with availability or self-administration of the drug can evoke intense subjective feelings of craving and can trigger episodes of relapse in abstinent cocaine abuse patients.
Experimental studies of drug-seeking behavior associated with drug-related stimuli in rats indicate that incentive motivational stimuli associated with cocaine can elicit and maintain robust cocaine-seeking behavior in the absence of drug availability. For example, rats responding for presentation of conditioned stimuli previously paired with food or cocaine showed a strong shift in preference for a cocaine- over a food-associated stimulus after receiving a noncontingent "priming" injection of cocaine. This effect was particularly sensitive to reversal by a dopamine D1 antagonist, implicating activation of D1 receptors in the motivational effects of cocaine under these conditions.
In rats trained to self-administer cocaine intravenously, presentation of a discriminative stimulus previously predictive of cocaine availability elicited significant and persistent responding after extended periods of abstinence and increased DA efflux in the NAc. The reinstatement of cocaine-seeking behavior was blocked by both dopamine D1 and D2 antagonists. Together, these observations implicate activation of dopaminergic mechanisms in the motivational effects of drug-associated environmental stimuli and drug-priming. Moreover, these data suggest that cocaine-related cues may exert a "priming" action since, like cocaine, these stimuli increase extracellular levels of DA in the NAc.
Summary
It has been proposed that drug addiction is the result of neuroadaptive processes within the central nervous system that oppose the acute reinforcing actions of drugs of abuse (Koob and Bloom 1988), leading to impairment in the mechanisms that mediate positive reinforcement and the emergence of affective changes such as anxiety, dysphoria, and depression during withdrawal. The results reveal perturbations in DA and 5-HT transmission in the NAc-neurochemical systems that are activated by cocaine self-administration and are deficient during withdrawal-as potential substrates for these affective changes. In addition, the results implicate neuroadaptive changes in extrahypothalamic CRF neurons and other brain stress circuitries in the motivational effects of psychostimulant withdrawal. Finally, it appears that environmental cues that become conditioned to the positive reinforcing effects of cocaine can mimic the pharmacological effect of this agent and, thereby, can initiate and maintain cocaine-seeking behavior.
Acknowledgment
This research was supported by National Institute on Drug Abuse Grant No. DA-07348.
References
- Rodriguez de Fonseca, F.; Carrera, M.R.A.; Navarro, M.; Koob, G.F.; and Weiss, F. Activation of corticotropin releasing factor in the limbic system during cannabinoid withdrawal. Science 276:2050 -2054, 1997.
- Koob, G.F., and Bloom, F.E. Cellular and molecular mechanisms of drug dependence. Science 242:715- 723, 1988.
- Koob, G.F.; Markou, A.; Weiss, F.; and Schultei s, G. Opponent process and drug dependence: Neurobiological mechanisms. Semin Neurosci 5:351-35 8, 1993.
- Markou, A., and Koob, G.F. Postcocaine anhedonia: An animal model of cocaine withdrawal. Neuropsychopharmacolog y4:17-26, 1991.
- Markou, A., and Koob, G.F. Bromocriptine reverses the elevation in intracranial self-stimulation thresholds observed in a rat model of cocaine withdrawal. Neuropsychopharmacolog y 7:213-22 4, 1992.
- Pich, E.M.; Lorang, M.; Yeganeh, M.; Rodriguez de Fonseca, F.; Raber, J.; Koob, G.F.; and Weiss, F. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci 15:5439- 5447, 1995.
- Parsons, L.H.; Kerr, T.M.; Weiss, F.; and Koob, G.F. Serotonin -1B receptor stimulation enhances cocaine reinforcement: Behavioral and neurochemical studies in rats. J Neurosci, submitted.
- Parsons, L.H.; Koob, G.F.; and Weiss, F. Serotonin dysfunction in the nucleus accumbens of rats during withdrawal after unlimited access to intravenous cocaine. J Pharmac ol Exp Ther 274:1182 -1191, 1995.
- Richter, R.M.; Pich, E.M.; Koob, G.F.; and Weiss, F. Sensitization of cocaine-stimulated increase in extracellular levels of corticotropin-releasing factor from the rat amygdala after repeated administration as determined by intracranial microdialysis. Neurosci Lett 187:169- 172, 1995.
- Richter, R.M., and Weiss, F. In vivo CRF release in rat amygdala is increased during withdrawal after cocaine self-administration with unlimited access. J Neurosci, submitted.
- Weiss, F.; Hurd, Y.L.; Ungerstedt, U.; Markou, A.; Plotsky, P.M.; and Koob, G.F. Neurochemical correlates of cocaine and ethanol self-administration . In: The Neurobiology of Drug and Alcohol Addiction. Kalivas, P.W., and Samson, H.H., eds. Ann N Y Acad Sci 654:220- 241, 1992a.
- Weiss, F.; Imperato, A.; Casu, M.A.; Mascia, M.S.; and Gessa, G.L. Opposite effects of stress on dopamine release in the limbic system of drug-naive and chronically amphetamine-treated rats. Eur J Pharmacol 337:219- 222, 1997.
- Weiss, F.; Markou, A.; Lorang, M.T.; and Koob, G.F. Basal extracellular dopamine levels in the nucleus acumbens are decreased during cocaine withdrawal after unlimited- access self-administration . Brain Res 593:314- 318, 1992b.
- Weiss, F.; Parsons, L.H.; and Markou, A. Neurochemistry of cocaine withdrawal. In: The Neurobiology of Cocaine: Cellular and Molecular Mechanisms. Hammer, Jr., R.L., ed. Boca Raton, FL: CRC Press, 1995. pp. 163-180.
- Wise, R.A. Neurobiology of addiction. Curr Opin Neurobiol6:243-251, 1996.
- Sensitization to Cocaine is Associated with Modified Calcium Transduction and Gene Expression in the Nucleus Accumbens
-
Peter W. Kalivas, Ph.D.
Washington State University
Pullman, WABehavioral sensitization induced by repeated cocaine administration is associated with long-term alterations in neurotransmission in the nucleus accumbens (Pierce and Kalivas 1997a). Notably, both presynaptic and postsynaptic dopamine transmissions are enhanced in cocaine-sensitized rats.
Recent data collected primarily by Margaret Gnegy at the University of Michigan, as well as by our own group, demonstrate that the increased releasability of dopamine in the nucleus accumbens or striatum of cocaine- or amphetamine-sensitized rats arises from an increase in calcium transduction. Specifically, there is an enduring increase in CaM-KII activity and the CaM-KII phosphorylated form of synapsin I in rats treated with repeated amphetamine (Pierce and Kalivas 1997b). The increased release of dopamine is antagonized by calcium channel blockers and CaM-KII inhibitors (Iwata et al. 1996). Furthermore, microinjection of these antagonists into the nucleus accumbens inhibits the expression of behavioral sensitization to cocaine (unpublished observations).
In addition to increased releasability of transmitter, repeated psychostimulant administration alters postsynaptic dopamine transduction in the nucleus accumbens, as evidenced by increased electrophysiological responsiveness to D1 dopamine receptor agonists (Henry and White 1991).
More recently, a number of laboratories have shown that repeated psychostimulant administration produces a change in gene expression. However, to date only two changes in gene expression parallel the time course of behavioral sensitization by enduring for weeks after discontinuing repeated drug administration. Both gene products are apparently transcription factors including a long-lasting AP-1 DNA binding complex (probably related to persistent expression of delta-FosB-related protein) (Nye et al. 1995) and NAC-1 (Cha et al. 1997). Recently, FosB-mutant mice were shown to have a heightened behavioral sensitivity to contingent and noncontingent cocaine administration (Hiroi et al. 1997). NAC-1 is a recently isolated mRNA that contains a POZ binding domain and is upregulated for at least 3 weeks after discontinuing repeated cocaine administration (Cha et al. 1997). Similar to the FosB mutant mice, reducing NAC-1 gene expression by microinjecting antisense oligonucleotide into the nucleus accumbens dramatically augmented the stimulant effects of acute and repeated cocaine administration (unpublished data). The fact that the inhibition and abolition of NAC-1 and FosB gene expression, respectively, enhanced the behavioral effects of cocaine indicates a possible role for either protein in both the acute rewarding and psychostimulant effects of cocaine, as well as in long-term behavioral alterations such as paranoia and drug- craving.
Acknowledgment
This research was supported by National Institute on Drug Abuse Grant No. DA-03906.
References
- Cha, X-Y.; Pierce, R.C.; Kalivas, P.W.; and Mackler, S.A. NAC-1, a rat brain mRNA, is increased in the nucleus accumbens three weeks after chronic cocaine self-administration. J Neurosci 17:6864-6871, 1997.
- Henry, D.J., and White, F.J. Repeated cocaine administration causes persistent enhancement of D1 dopamine receptor sensitivity within the rat nucleus accumbens. J Pharmacol Exp Ther 258:882-890, 1991.
- Hiroi, N.; Brown, J.R.; Haile, C.N.; Ye, H.; Greenberg, M.E.; and Nestler, E.J. FosB mutant mice: Loss of chronic cocaine induction of Fos-related proteins and heightened sensitivity to cocaine's psychomotor and rewarding effects. Proc Natl Acad Sci U S A 94:10397-10402, 1997.
- Iwata, S.; Hewlett, G.H.; Ferrell, S.T.; Czernik, A.J.; Meiri, K.F.; and Gnegy, M.E. Increased in vivo phosphorylation state of neuromodulin and synapsin I in striatum from rats treated with repeated amphetamine. J Pharmacol Exp Ther278:1428-1434, 1996.
- Nye, H.E.; Hope, B.T.; Kelz, M.B.; Iadorola, M.; and Nestler, E.J. Pharmacological studies of the regulation of chronic FOS-related antigen induction by cocaine in the striatum and nucleus accumbens. J Pharmacol Exp Ther 275:1671-1680, 1995.
- Pierce, R.C., and Kalivas, P.W. A circuitry model of the expression of behavioral sensitization to amphetamine-like psychostimulants. Brain Res Brain Res Rev 25:192-216, 1997a.
- Pierce, R.C., and Kalivas, P.W. Repeated cocaine modifies the mechanism by which amphetamine releases dopamine. J Neurosci 17:3254-3261, 1997b.
- CART: A Novel Addiction Peptide
-
Michael J. Kuhar, Ph.D.
Emory University
Atlanta, GACART (cocaine- and amphetamine-regulated transcription factors) peptides are novel putative neurotransmitters/cotransmitters. They have psychostimulant-like effects in that they alter locomotor activity and feeding behaviors. CART peptides are localized to regions in the brain that are associated with these behaviors, as well as other regions suggesting involvement in a variety of physiologic functions.
Acknowledgment
This research was supported by National Institute on Drug Abuse Grant No. DA-10732.
Selected References
- Couceyro, P.R.; Koylu, E.O.; and Kuhar, M.J. Further studies on the anatomical distribution of CART by in situ hybridization. J Chem Neuroanat 12:229-241, 1997.
- Douglass, J.; McKinzie, A.A.; and Couceyro, P. PCR differential display identifies a rat brain mRNA that is transcriptionally regulated by cocaine and amphetamine. J Neurosci 15:2471-2481, 1995.
- Koylu, E.O.; Couceyro, P.R.; Lambert, P.D.; Ling, N.C.; DeSouza, E.B.; and Kuhar, M.J. Immunohistochemical localization of novel CART peptides in rat hypothalamus, pituitary and adrenal gland. J Neuroendocrinol 9:823- 833, 1997.
- Lambert, P.D.; Couceyro, P.R.; Koylu, E.O.; Ling, N.C.; DeSouza, E.B.; and Kuhar, M.J. CART peptides have a physiological role in the central control of food intake (Abstract). Abstracts of the 27th Annual Meeting of the Society for Neuroscience, New Orleans, LA, Oct 25-30, 1997. p. 966.
- Smith, Y.; Koylu, E.O.; Couceyro, P.; and Kuhar, M.J. Ultrastructural localization of CART (cocaine- and amphetamine-regulated transcript) peptides in the nucleus accumbens of monkeys. Synapse 27:90-94, 1997.